
Installation and data preparation
The detailed instruction on VRPG's installation and data preparation can be found at its GitHub repository.
Graphical user interface
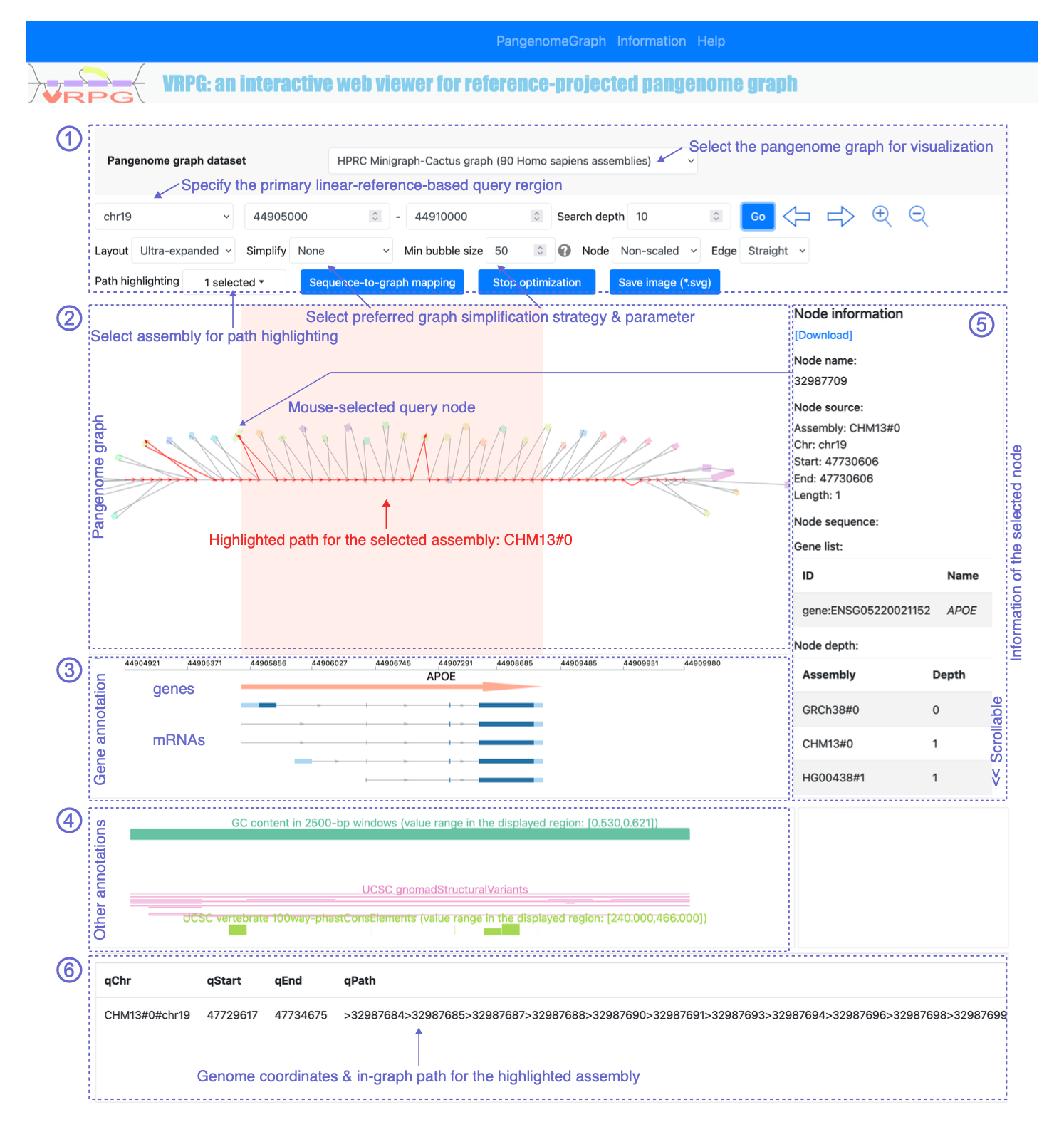
Figure 1. The interactive user interface of VRPG when opened in a web browser.
The control panel
Select 'Pangenome graph dataset'
Users can select their interested pangenome graph dataset to visualize from the list provided here, which was defined by the user who deployed the VRPG the web application (see the installation and data preparation section for details)
Specify the Query genome coordinates along the primary linear reference genome.
The chromosome/scaffold/contig names can only be selected from a pre-defined list, which was generated by VRPG's data preparation submodule and loaded into VRPG's web application automatically. The start and end coordinates should be integer without any flanking space.
VRPG uses web browser to render the graph. When too many nodes are loaded to the browser, the browser may fail to response. In practice, we recommended to limit the size of the queried genomic region to 1 Mb, so that the browser can respond within a few milliseconds.
Specify 'Search depth'
A positive integer without any flanking space should be provided here (Default: 10). Lower this value may reduce the complexity of the subgraph but at the cost of displaying incomplete graph bubble. VRPG implemented a breadth first algorithm to extract the subgraph from the indexed graph data. Each node along the primary linear reference within the user defined region is taken as a starting point for the search.
During the search, if there is no additional non-reference node or the search depth is greater than the specified Search depth value, the search will be terminated. Such programed behavior is also conditioned on the index depth value specified during VRPG's graph index building stage, which imposed an upper bound for the maximal search depth. When the specified search depth value is greater than this value, the search depth will be automatically reset to the index depth behind the scene. For the graphs hosted in this demonstration site, their graph index depths were set to 100.

Figure 2. The effect of different search depth values on graph visualization. (A) Search depth set as 10. (B) Search depth set as 2.
Select 'Layout'
Five optional layouts are provided: Ultra expanded (default), Expanded, Squeezed, Hierarchical expanded, Hierarchical squeezed. For the Ultra expanded, Expanded, and Squeezed layouts, all reference-matching nodes are always scaled and displayed along the center line, while nodes that represent genomic variants relative to the primary linear reference are shown alongside, which can be optionally scaled or not. For the 'Hierarchical-expanded' and 'Hierarchical-squeezed' layouts, the reference-matching nodes are placed along a straight line on the top and all non-reference nodes are placed underneath. Nodes with higher ranks (as defined in the rGFA format) are placed higher than nodes with lower ranks. Normally, when the visualized subgraph topology is relatively simple, the 'Ultra expanded', 'Expanded', and 'Hierarchical-expanded' layouts are recommended. Otherwise, the 'Squeezed' or 'Hierarchical-squeezed' layouts are more likely to work better. In addition, users can also manually drag the node with the mouse for further layout fine-tunning.

Figure 3. The effect of different layout choices on graph visualization. (A) With the 'Ultra expanded' layout. (B) With the 'Squeezed' layout. The human HLA region (chr6:29940912-30009956) in the HPRC Minigraph-Cactus Pangenome is visualized here.
Apply 'Simplify'
Three graph simplification strategies are provided for layout simplification: 'Nonref nodes' (default), 'All nodes', and 'None'. These simplification strategies could be helpful when visualizing genomic regions with complex graph topologies. Here 'Nonref nodes' means only simplify the non-reference-matching nodes by trimming off those bubbles (See Define 'Min bubble size' below') representing for very short genomic segment (e.g., <50 bp). Here a bubble refers to the circular structure in a pangenome graph and it contains at least two nodes. A bubble in a pangenome graph generally indicates genomic variants. 'All nodes' means simplify both non-reference-matching and reference-matching nodes by trimming off those bubbles representing for very short genomic segment (e.g., <50 bp). 'None' means no simplification. Note that when the 'All nodes' simplification strategy is selected, the annotation tracks will not be correctly displayed.

Figure 4. The effect of applying different simplification strategies on graph visualization. (A) Simplification strategy as 'None'. (B) Simplification strategy as 'Nonref nodes'. (C) Simplification strategy as 'All nodes'.
Define 'Min bubble size'
We define the 'Min bubble size' as minimal approximated cumulative node length for bubbles to be displayed. This parameter should be used together with the above-mentioned layout simplification option.
Select display style for 'Node'
The primary linear-reference-matching nodes are always displayed in scale, with the node length proportional to the actual sequence length. This option is mainly used to control the display style of the non-reference-matching nodes. When 'Scaled' is selected, the non-reference nodes will also be approximately scaled. In the Nonscaled style (default), the non-reference node length will be automatically determined by the d3-force (https://d3js.org/d3-force) layout. Nodes in the rendered pangenome graph are illustrated in different colors, among which those derived from the same assembly will be rendered with the same color. When clicking on a node, the selected node will be thickened, and its corresponding information will be reported in the 'Node information' panel.
Select display style for 'Edge'
Graph edges can be displayed as straight lines (default) or curved lines. When clicking on a graph edge, the edge will be colored in re and its color will revert to black on a second click.
Define 'Path highlighting'
User can select one or more assemblies for 'Path highlighting' in the rendered graph layout. Click on the 'Path highlight' checkbox for assembly selection and click it again to hide the check box. One or more assemblies can be selected at once. Upon assembly selection is finished, click on the 'GO' button shown on the right for rendering the highlighted graph. The traversing paths of the selected query assembly (-ies) will be marked in the graph, with all matched nodes and edges highlighted in red. The arrows on the matched edges reflect the traversing direction of the highlighted linear genome assemblies in the pangenome graph. When a single assembly is selected for path highlighting, the in-node paths will also be thickened on a two-state basis (thin vs. thick) if the highlighted genome assembly traverses the corresponding node(s) more than once, which implies the occurrence of genomic duplications. Also, the corresponding path information will be reported in the Path information panel. When multiple assemblies are selected for path highlighting, their paths can still be highlighted in red but these paths will not be thicken in the case of genomic duplications and their path information will not be reported.
Apply 'Sequence-to-graph mapping'
The Sequence-to-graph mapping is powered by Minigraph. For the time being, this function is only supported for pangenome graphs built by Minigraph, but we will work on extending this function to graphs built by Minigraph-Cactus and PGGB in the near future. By clinking on the 'Sequence-to-graph mapping' button, a search box will show up for the users to specify the query sequence (in FASTA format). For the demonstration site, we imposed a query sequence length limit of 1Mb. If a search hit can be found in the pangenome graph, a task ID and a list of matched primary linear reference genome coordinates will show up in the form of refChr, refStart, and refEnd. This match primary reference coordinates could either come from a direct match or an indirect match, which is the matched peripheral nodes together with its residing bubble being further projected back to the primary reference genome. Users can further visualize the matched subgraph by checking the 'View mapping results' box, which is right above the 'Task ID'. And then re-specify the VRPG query coordinates using the hit coordinates on the primary reference and then click the 'Go' button. The hit sequence will be shown in the resulting sub-graph with the hit path further highlighted. The detailed mapping information (e.g., matched query start, matched query end, matched path, and matched CIGAR string, etc.) will be further reported in the Path information panel. If a match has been found but the highlighted sequence can't be seen in the corresponding sub-graph, one can increase the 'Search depth' value described earlier in this manual. Once the 'Sequence-to-graph mapping' analysis is done, please remember to uncheck the 'View mapping results' box, so that the 'Path highlighting' function will not be affected.

Figure 5. The effect of applying sequence-to-graph mapping and hit path highlighting. (A) Specifying the genome coordinates based on the matched region on the primary reference. (B) The resulting visualization of the sequence-to-graph mapping hit (marked by the blue box) in the pangenome graph.
Apply 'Stop optimization'
This option is for users to manually stop the layout optimization when the process takes too long.
Save image
Click the 'Save image' button to save the rendered subgraph along with the associated genomic annotations into an image file (SVG format).
Mark the genic region
When click on the genic region of a gene shown in the gene annotation track, its gene name will show up. Meanwhile, a rectangular block marking the corresponding genic region will be displayed as well. In this state, the graph nodes and edges will not be selectable due to the graphic layer order restriction. To make the selection, please click on the genic region again to cancel the marked block, after which nodes and edges will become selectable again.
Obtain 'Node information'
The reported information includes the assembly and genomic coordinates source of the selected node, a list of annotated genes covered by this node, and estimated copy numbers of the node-representing genomic sequence in all graph constituent assemblies. The Start and End positions are 1-base coordinates. The node depth for each assembly is calculated by aligning the corresponding assembly to the pangenome graph. A depth of 1 means the query assembly has a unique match with the node. A depth of 0 means the corresponding node is absent from the query assembly. A depth of 2 or more means the query assembly has 2 or more copies of the node. Note that for the demonstration site, we have not storage the node sequence for the 3 human pangenome graphs given the storage limitation of our webserver. Therefore, when clicking on a node in these graphs on our demonstration site, the reported node sequence will be an empty string.
Obtain 'Path information'
Upon assembly-to-graph path highlighting or sequence-to-graph mapping, the matched query assembly/sequence coordinates and in-graph path will be reported in the 'Path information' panel. For GFAv1-formatted input pangenome graph, the path information reported here should include 'qChr', 'qStart', 'qEnd' and 'qPath'. For rGFA-formatted input pangenome graph, the path information reported here should further include 'qCigar' in addition to 'qChr', 'qStart', 'qEnd' and 'qPath'. Here 'qChr' means the name of the matched query sequence (for 'Sequence-to-graph mapping') or the name of the matched chromosome/scaffold/contig in the selected assembly (for 'Path highlighting'). 'qStart' means the 1-based starting coordinate of the matched query sequence (for 'Sequence-to-graph mapping') or matched assembly chromosome/scaffold/contig (for 'Path highlighting'). 'qStart' means the 1-based ending coordinate of the matched query sequence (for 'Sequence-to-graph mapping') or matched assembly chromosome/scaffold/contig (for 'Path highlighting'). 'qPath' means the hit path of the query in the pangenome graph. Here we denoted the qPath in rGFA-style with '>' and '<' indicate the positive and negative strand respectively. 'qCigar' is the CIGAR-string denote the quality of the sequence alignment. The notation of '>' and '<' in 'qCigar' the same as decribed for 'qPath'. They are introduced here as the alignment splitting record along different matched individual nodes. Other than these introduced '>' and '<' signs, the qCigar string shown in the Path information panel should be equivalent to the CIGAR string recorded in the GAF file generated by Minigraph.

Figure 6. The Path information for a highlighted path in the graph. (A) This is an example in the yeast pangenome graph, in which the path corresponding to BAD#collapsed was highlighted (in red). (B) The corresponding report in the Path information panel. We can see that multiple chromosomes (e.g., chrI, chrIV, etc.) of the BAD#collapsed assembly have been mapped to the pangenome grpah in the visualized region. Taking the first alignment as an example, it means the BAD#collapsed#chrI:6784-16047 matched with the visualized pangenome graph, with the exact node-matching order defined in qPath, i.e., from node 1 to node 29702, to node 3, …, to node 14, to node 15. The first '>' in column qCigar corresponds the first '>' in qPath. '35=' means 35 bases of the query sequence can match with the sequence of node 1. Users can check the CIGAR specification to decode the following string by themselves when needed.
Coordinate
The displayed genome coordinates and feature annotation tracks are fully scaled with the pangenome graph horizontally. To make this possible, the annotation tracks are dynamically stretched or shrunk along with the displayed graph to make them fully synchronized.

Figure 7. Synchronized genome coordinate system of the pangenome graph and feature annotation tracks. Take the human OR4F5 gene region as an example. The vertical dashed lines y1 and y6 indicate the boundaries of the gene OR4F5, while y2, y3, y4 and y5 indicate the boundaries of two adjacent graph nodes overlapped with the ORF4F5 gene.
Genomic variants
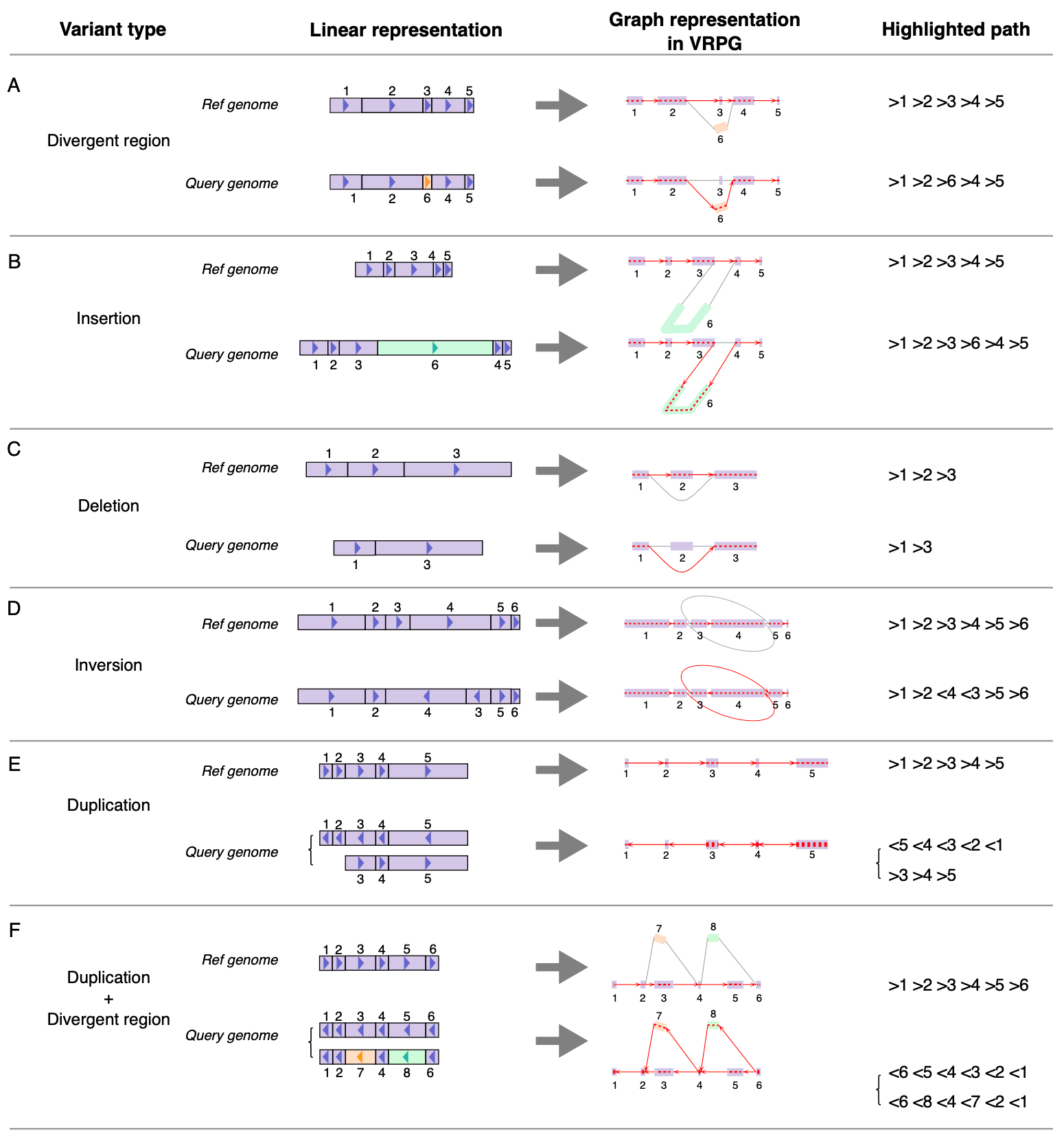
Figure 8. VRPG visualization for different types of genomic variants. For different types of genomic variants such as (A) Divergent region, (B) Insertion, (C) Deletion, (D) Inversion, (E) Duplication, and (F) Duplication with divergent region, their graph representations in VRPG are depicted with path highlighting for the reference and query genome respectively. Information of the highlighted path regarding nodes' traversing order and orientation is further reported (in rGFA-specified style) in VRPG's the path information panel.
Naming scheme of input assemblies
The naming scheme of input assemblies in VRPG follows the PanSN prefix naming pattern in general, although with a bit relaxation. The assembly name should consist of three parts: sample tag, delimiter, and haplotype tag. For our demonstration cases, we used "#" as the delimiter. Regarding the haplotype tag, for the yeast reference pangenome graph, we used "HP0" to denote haplotypes of haploid or homozygous diploid strains, while using "collapsed", "HP1", and "HP2" to denote collapsed, or the two phased haplotypes of heterozygous diploid strains. For Mingraph graph, "0" was used to denote haplotypes of haploid or collapsed samples while "mat" and "pat" were used to denote the maternal and paternal haplotypes of the phased diploid samples. For Minigrpah-Cactus and PGGB graphs “1” and “2” were used to denote the two phased haplotypes.
Questions and Answers (Q & A) on VRPG's algorithmic design
Q1: Is there a maximal size limit for the query region when using VRPG for graph visualization?
R1: In practice, we recommend limiting the query region size to 1 Mb to restrain the total number of nodes and edges for rendering, so that the web browser will not get overwhelmed during graph rendering.
Q2: Does the block indexing scheme implemented by VRPG actually guarantee that all potentially relevant material is retrieved, or is it heuristic?
R2: VRPG uses the '--xDep' parameter (specified via vrpg_preprocess.py or gfa2view) to set the hard maximal search depth allowance for graph traversing during block indexing. When setting this value large enough (default: 10), VRPG should be able to exhaustively retrieve all potentially relevant material for rendering. Note that another 'search depth' option (default: 10) is also provided to set the soft maximal search depth allowance for the final graph rendering, which is inherently constrained by the hard limit set by '--xDep'.
Q3: In VRPG's graph block indexing and subgraph rendering, will the blocks that are not actually in the query region but instead are connected to the query region by edges also be considered?
R3: When asking for a query region, VRPG will locate and render all blocks that are either directly overlapped with the query region (based on the primary linear reference cooridnates) or connected to the query region within the abovementioned soft and hard maximal search depth allowances.
Q4: What happens to material that was on the path between contigs when the inter-chromosomal paths are cut?
R4: In this case, the eventual primary-reference-based chromosome assignment will depend on the specified soft and hard maximal search depth allowance. If the connecting paths to both chromosomes are within the search depth allowance, this material will be assigned to both chromosomes. If only one of the two chromosome is within the search depth allowance, the material will be assigned to this chromosome. If neither chromosome is within the search depth allowance, the material will not be assigned to any of the two chromosomes.
Q5: What happens to material that is equally closely connected to primary reference nodes in multiple blocks of the same contig?
R5: In this case, the material will be found and stored in each individual block. This may cause redundancy, but it guarantees the discovery of all non-primary-reference nodes that can be connected to the primary reference.
Q6: Will VRPG use the rank information (i.e., the SR tag) provided in the rGFA file?
R6: When the input graph is in the rGFA format (https://github.com/lh3/gfatools/blob/master/doc/rGFA.md), a primary-linear reference coordinate system should have been predefined, with all nodes from this linear reference been labeled as rank-0 (defined with the SR tag). Those incrementally added non-linear-reference nodes derived from other graph-constituent assemblies are labeled with lower ranks (e.g., 1, 2, etc). VRPG can readily use this information for organizing the displayed graph layouts during graph visualization. In addition, Minigraph is further used to align each graph-constituent-assembly to the rGFA-graph to establish the assembly-to-graph path tracing information.
Q7: How does VRPG use the path information recorded in the GFA file?
R7: When the input graph is in the GFA format (https://github.com/GFA-spec/GFA-spec/blob/master/GFA1.md), VRPG's graph2view module will extract the path (the P-line) and walk (the W-line) information recorded in the GFA format to build the primary-linear-reference-based coordinate system of the pangenome graph. During this process, VRPG will transform the original pangenome graph into a linear-reference-projected pangenome graph with conditionally inserted shadow nodes and edges (for collapsed nodes) for establishing the entire linear coordinate system for user-specified linear reference genome. Meanwhile, the predefined path information in the GFA file will also be used for directly tracing the assembly-to-graph path of each graph-constituent assembly.
Q8: How many annotation tracks and how many annotation records within each track can VRPG display?
R8: The theoretical maximal number of annotation tracks are 65535 for VRPG. In practice, we recommend a maximum of 10 tracks for legibility consideration. For each track, the maximal number of annotation records allowed is 50 by default, which can be further adjusted by specifying the '--layer' option for the 'GraphAnno addBed' command in VRPG.
Contact
The VRPG software and the associated web server are developed by Evomics Lab at Sun Yat-sen University Cancer Center (SYSUCC). If you have any questions, please feel free to contact us via yuejiaxing[at]gmail[dot]com.